
- Як проявляється хвороба фенілкетонурія
- Механізм розвитку захворювання
- Фенілкетонурія у дітей
- симптоми захворювання
- Причини і провокуючі фактори
- діагностика
- Лікування класичної фенілкетонурії
- Особливості харчування новонароджених і дієтотерапія
- Дієта для дітей дошкільного віку та школярів
- Групи продуктів при ФКУ
- Як контролювати рівень фенілаланіну в крові
- Відео
Допоможіть розробці сайту, ділитися статтею з друзями!
Захворювання, виникнення якого пов'язане з вадами в генетичному клітинному апараті, - фенілкетонурія - входить в нечисленний перелік спадкових хвороб, що піддаються лікуванню. Першовідкривачем цієї недуги був лікар з Норвегії І.А. Феллінг, пізніше було виявлено, що за розвиток і перебіг хвороби відповідає єдиний ген, іменований геном фенілаланінгідроксилази (довге плече дванадцятий хромосоми, що містить до 4, 5% всього матеріалу ДНК клітини). Наслідуваний дефект призводить до часткової або повної деактивації ферменту печінки фенілаланін-4-гідроксилази.
Як проявляється хвороба фенілкетонурія
Спадкове захворювання фенілкетонурія (ФКУ) призводить до хронічного отруєння організму токсичними речовинами, утворених внаслідок порушеного метаболізму амінокислот і процесу гідроксилювання фенілаланіну. Постійна інтоксикація викликає ураження центральної нервової системи (ЦНС), проявом чого виступає прогресуюче зниження інтелекту (фенилпировиноградная олігофренія).
Хвороба Фелінга проявляється в надмірному накопиченні в організмі фенілаланіну і продуктів його неправильного обміну. До інших чинників розвитку фенілкетонурії відноситься порушене транспортування амінокислот через гематоенцефалічний бар'єр, низька кількість нейротрансмітерів (серотонін, гістамін, допамін). При відсутності своєчасного лікування захворювання призводить до розумової відсталості і може стати причиною загибелі дитини.

Механізм розвитку захворювання
Прічінообразующім фактором виникнення генних порушень є метаболічний блок, який перешкоджає утворенню фенілаланін-4-гідроксилази (фермент, який відповідає за перетворення амінокислоти фенілаланіну в тирозин). Протеіногенних амінокислота тирозин є складовою частиною білків і пігменту меланіну, тому є необхідним елементом для функціонування всіх систем організму, а її нестача призводить до ферментопатии.
Наслідком придушення освіти метаболіти, викликаного мутаційної інактивацією ферменту, є активування допоміжних шляхів обміну фенілаланіну. Ароматична альфа-амінокислота в результаті дефектних обмінних процесів розпадається на токсичні похідні, які в нормальних умовах не утворюються:
- фенілпіровиноградну кислоту (фенілпіруват) - жирно-ароматична альфа-кетокислот, її освіту призводить до мієлінізації відростків нейронів і недоумства;
- фенілмолочная кислоту - продукт, що утворився при відновленні фенилпировиноградной кислоти;
- фенілетиламін - початкова з'єднання для біологічно активних передавачів електрохімічних імпульсів, підвищує концентрацію дофаміну, адреналіну і норадреналіну;
- ортофенілацетат - токсична речовина, що викликає порушення обмінних процесів жироподібних сполук в головному мозку.
Медична статистика свідчить про те, що патологічно змінений ген присутній у 2% населення, але при цьому він ніяк не проявляє себе. Генетичний дефект передається дитині від батьків тільки при наявності захворювання у обох партнерів, при цьому немовля в 50% випадків стає носієм гена, що мутує, залишаючись здоровим. Імовірність того, що фенілкетонурія у новонароджених призведе до захворювання, становить 25%.
За яким типом успадковується
Хвороба Фелінга є генетичним відхиленням, що успадковується по аутосомно-рецесивним типом. Такий тип спадкування означає, що розвиток ознак вродженого захворювання станеться тільки при спадкуванні дитиною по одній дефектної копій гена від обох батьків, які є гетерозиготними носіями видозміненого гена.
Розвиток вродженого захворювання в 99% випадків викликає мутація гена, відповідального за кодування ферменту, що забезпечує синтез фенілаланін-4-гідроксилази (класична фенілкетонурія). До 1% генетичних захворювань пов'язані з мутаційними змінами, що відбуваються в інших генах, і викликають недостатність дігідроптерідінредуктази (ФКУ II типу) або тетрагідробіоптеріна (ФКУ III типу).
Фенілкетонурія у дітей
Класична форма генетичного захворювання у дітей в більшості випадків виявляється в зовнішньо помітних ознаках, починаючи з 3-9 місяця життя. Новонароджені, що мають дефектний ген, виглядають здоровими, відмінною рисою буває специфічний хабітус (зовнішній вигляд) дитини. Виражена симптоматика з'являється через 6-12 місяців після народження.
ФКУ II типу характеризується тим, що перші клінічні симптоми з'являються через 1, 5 року з моменту появи на світ. Ознаки захворювання не зникають після діагностування генетичних відхилень і початку дієтотерапії. Цей вид вродженої хвороби нерідко призводить до летального результату на 2-3 році життя дитини. Найчастішими симптомами ФКУ II типу є:
- виражені відхилення в розумовому розвитку;
- гиперрефлексия;
- порушення рухових функцій всіх кінцівок;
- синдром безконтрольних м'язових скорочень.
- висока ступінь розумової відсталості;
- явно зменшений розмір черепа по відношенню до інших частин тіла;
- спастичність мускулатури (при цьому можлива повна знерухомлених кінцівок).

Прояви хвороби Фелінга
В ході клінічних досліджень і спостережень були висунуті припущення, що вплив токсичних похідних фенілаланіновой обміну викликає зниження інтелектуальних здібностей, яке носить прогресуючий характер і може призвести до недоумства (олігофренії, ідіотії). Серед передбачуваних причин незворотних порушень мозкової діяльності самої обґрунтованої вважається викликана зниженням рівня тирозину брак нейромедіаторів, що передають імпульси між нейронами.
Точну причинно-наслідковий зв'язок між спадковим захворюванням і порушеннями мозку до теперішнього часу не виявлено, як і механізм розвитку внаслідок фенілкетонурії таких психічних станів, як ехопраксія, ехолалія, нападів гніву і дратівливості. Дані результатів аналізів свідчать про те, що фенілаланін діє токсично на мозок, що теж може викликати зниження інтелекту.
Статура і фенотипічні особливості
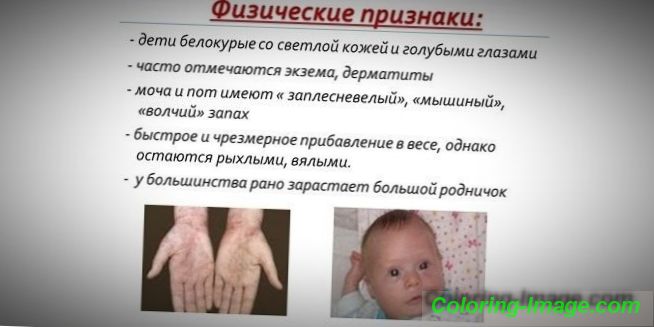
З огляду на те, що насиченість пігментом шкіри і волосся залежить від рівня тирозину в мітохондріях гепатоцитів, а фенілкетонурія призводить до зупинки конверсії фенілаланіну, пацієнти з цим захворюванням мають фенотипічні особливості (рецесивні ознаки). Підвищений м'язовий тонус стає причиною появи відхилень у будові тіла - воно стає диспластичне. До відмінних зовнішніми ознаками фенілкетонурії відносяться:
- гипопигментация - світла шкіра, блідо-блакитні очі, знебарвлені волосся;
- синюшність кінцівок;
- зменшений розмір голови;
- специфічне положення тіла - при спробах стояти або сидіти дитина приймає позу «кравця» (руки і ноги зігнуті в суглобах).
симптоми захворювання
При своєчасному виявленні хвороба Фелінга піддається успішному лікуванню шляхом коригування харчування, і розвиток дитини відбувається відповідно його віковій групі. Труднощі виявлення генної мутації полягає в тому, що ранні ознаки важко виявити навіть досвідченому педіатра. Виразність симптоматики вродженого захворювання посилюється в міру дорослішання дитини, тому що вживання білкової їжі сприяє розвитку порушень ЦНС.
Ознаки у новонароджених
Протягом перших днів життя дитини ознаки патологічних відхилень виявити важко - малюк поводиться природно, затримки в розвитку не спостерігається. Симптоми захворювання вперше починають проявлятися через 2-6 місяців після народження. Батьків має насторожити поведінку малюка, яке характеризується низькою активністю, млявістю, або, навпаки, занепокоєнням, гіперзбудливості.
З початком грудного вигодовування в організм новонародженого з молоком починають надходити білки, що служить каталізатором появи перших ознак, однозначно свідчать про те, що захворювання початок прогресувати. До специфічних клінічних проявів хвороби відносяться:
- постійна блювота (часто приймається за вроджене звуження воротаря);
- часте зригування;
- відсутність реакції на зовнішні подразники;
- м'язова дистонія (знижене напруга м'язів);
- судомний синдром (судоми епілептичного або неепілептичного характеру).
Симптоми у дітей після 6 місяців
Якщо маніфестація генетичної хвороби не відбулася (або не була помічена) протягом перших 6 місяців з моменту народження дитини, то після цього періоду вже можна точно визначити відставання в психомоторному розвитку. Симптомами генетичних порушень, викликаних ферментним дефіцитом, у дітей, старше півроку, є:
- зниження активності (аж до повної байдужості);
- відсутність спроб до самостійного вставання, сидіння;
- особливий «мишачий» запах шкіри (запах цвілі виникає внаслідок виведення токсичних похідних фенілаланіну через потові залози і сечу);
- втрата здатності до візуального розпізнавання осіб батьків;
- лущення шкіри;
- поява дерматитів, екземи, склеродермії.
Прогресування захворювання при відсутності лікування в дитячому віці
Якщо відхилення в розвитку не були виявлені в дитячому віці, і відповідне лікування не проводилося, то захворювання починає активно прогресувати і нерідко призводить до інвалідності. Відсутність терапії на ранньому етапі хвороби викликає поява таких симптомів хвороби вже у віці 1, 5 років:
- мікроцефалія (зменшені розміри головного мозку);
- прогнатия (зміщення верхнього зубного ряду вперед);
- пізнє прорізування зубів;
- гіпоплазія емалі (витончення або повна відсутність зубної емалі);
- затримка мовного розвитку аж до повної відсутності мовлення;
- 3, 4 ступінь олігофренії (затримка психічного розвитку, розумова відсталість);
- вроджені вади серця (дефекти в структурі серцевого м'яза, відділах серця, великих судинах);
- розлади вегетативної системи (акроціаноз, підвищена пітливість, артеріальна гіпотонія);
- запори.
Причини і провокуючі фактори
Для прояви мутації з аутосомно-рецесивним характером успадкування дефектний ген повинен бути успадкований від обох батьків. Генетичні захворювання такого типу зустрічаються з однаковою частотою у новонароджених хлопчиків і дівчаток. Патогенез ФКУ зумовлюється порушенням обміну фенілаланіну, який може протікати в 3 формах. Лікуванню дієтотерапією піддається тільки класична фенілкетонурія I типу.
Атипові форми захворювання не можна вилікувати шляхом коригування харчування. Ці відхилення викликані дефіцитом тетрагідроптеріна, дегідроптерінредуктази (рідше - пірувоілтетрагідроптерінсінтази, гуанозин-5-тріфосфатціклогідролази і ін.). Більшість випадків летальних результатів зафіксовано серед хворих рідкісними варіаціями ФКУ, при цьому клінічні прояви всіх форм хвороби аналогічні. Ризик народження дитини з мутував геном фенілаланінгідроксилази зростає, якщо його батьки є близькими родичами (при близькоспоріднених шлюбах).
діагностика
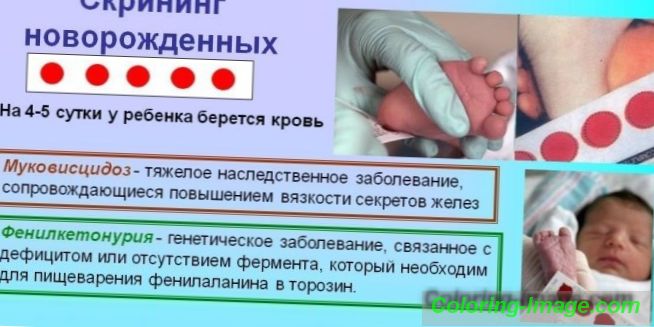
При підозрі на генетичні порушення діагноз встановлюється на підставі сукупності даних, отриманих в результаті вивчення анамнезу хвороби - генеалогічних відомостях, результати клінічних та медико-генетичних досліджень. Для своєчасного виявлення вроджених захворювань (ФКУ, муковісцидозу, галактоземії і ін.) Розроблена програма обов'язкового масового обстеження в лабораторних умовах всіх новонароджених дітей (неонатальний скринінг).
Якщо майбутні батьки знають про носійство гена, що мутує, сучасна медицина пропонує способи виявлення дефекту на етапі вагітності (допологова діагностика плоду інвазивних методом). Для поділу фенілкетонурії на види за ступенем тяжкості застосовується умовна класифікація, яка заснована на рівні фенілаланіну в безфібріногенной рідини, отриманої з плазми крові:
- Важка фенілкетонурія - 1200 мкмоль / л.
- Середня - 60-1200 мкмоль / л.
- Легка (не потребує лікування) - 480 мкмоль / л.
Скринінг-тест
Виявлення генетичних відхилень відбувається в кілька етапів. На першому етапі в пологовому будинку у всіх немовлят на 3-5 день життя здійснюється забір периферичної крові (з п'яти) для досліджень. Матеріал наноситься на паперовий бланк і відправляється в біохімічну лабораторію, де відбувається його біохімічний аналіз. На другому етапі скринінгу-тесту визначається відповідність концентрації фенілаланіну нормального значення.
Якщо патологічних змін не виявлено, діагностика завершується, про що робиться запис в картці дитини. При наявності відхилень від норми результати діагностики направляються лікаря-педіатра для забезпечення уточнюючого дослідження зразка крові новонародженого. Здоров'я малюка залежить від своєчасного і точного виконання всіх заходів по виявленню відхилень. Якщо діагноз підтвердиться після повторного скринінг-тесту, батьки дитини будуть спрямовані в клініку до дитячого генетику для призначення лікування.
Аналізи і дослідження для підтвердження діагнозу
Повторна діагностика при виявленні під час проведення первинного скринінг-тесту відхилень від норми здійснюється шляхом повторної здачі аналізів. Крім визначення вмісту фенілаланіну в крові до методів діагностики ФКУ у дітей і дорослих відносяться:
- проба Фелінга - визначення фенилпировиноградной кислоти в сечі шляхом додавання до біоматеріалів хлориду заліза (відбувається потемніння синьо-зелений колір);
- тест Гатрі - оцінка ступеня реакції мікроорганізмів на продукти обміну або ферменти, що містяться в крові пацієнта;
- хроматографія - вивчення хімічних властивостей речовин, розподілених між двома фазами;
- флуориметр - опромінення біоматеріалу монохроматическим випромінюванням для визначення концентрації містяться в ньому речовин;
- електроенцефалографія - діагностика електричної активності головного мозку;
- магнітно-резонансна томографія - збудження атомних ядер клітин електромагнітними хвилями і вимірювання їх відгуку.
Лікування класичної фенілкетонурії
В основі терапії фенілкетонурії лежить обмеження споживання продуктів, які є джерелом білків тваринного і рослинного походження. Єдиним способом успішного лікування є дієтотерапія, адекватність якої оцінюється за змістом фенілаланіну в сироватці крові. Гранично допустимий рівень амінокислоти у хворих різних вікових груп становить:
- у новонароджених і дітей до 3 років - до 242 мкмоль / л;
- у дошкільнят - до 360 мкмоль / л;
- у пацієнтів у віці від 7 до 14 років - до 480 мкмоль / л;
- у підлітків - до 600 мкмоль / л.
Від того, на якому етапі захворювання проведена корекція раціону, залежить ефективність дієти. При ранній діагностиці вродженої патології дієтотерапія призначається з 8 тижні життя (після цього періоду вже починаються незворотні зміни). Відсутність своєчасних заходів призводить до ускладнень і зниження рівня інтелекту на 4 бали за 1 місяць з моменту народження до початку лікування.

З огляду на те, що лікувальна дієта при фенілкетонурії передбачає повне виключення з раціону тваринного білка, з'являється необхідність застосування інших джерел необхідних амінокислот, а також вітамінів групи В, кальцій і фосфоровмісних мінеральних сполук. До продуктів, що призначається в якості добавок до безбілкову раціону, відносяться:
- білковий гідролізат (Аміго, Аміназол, Фібріносол);
- що не містять фенілаланін суміші, насичені незамінними амінокислотами - Тетрафен, Феніл-фрі.
Поряд з лікувальними заходами щодо усунення причини порушень функціонування організму слід проводити симптоматичне лікування, спрямоване на усунення дефектів мовлення і нормалізацію координації рухів. Комплексна терапія включає фізіотерапевтичні процедури, масаж, допомога логопеда, психолога, виконання гімнастичних вправ. У ряді випадку спільно з дієтотерапією показаний прийом антиконвульсантів, ноотропних і судинних препаратів.
Особливості лікування атипових форм
Фенілкетонурія II і III типу не піддається лікуванню низкобелковой дієтою - рівень фенілаланіну в крові залишається незмінним при обмеженні надходження білка в організм або клінічна симптоматика прогресує навіть при зниженні рівня амінокислоти. Ефективна терапія цих форм захворювання здійснюється з застосуванням:
- тетрагідробіоптеріна - фактора ураженого ферменту;
- синтетичних аналогів тетрагідробіоптеріна - ці речовини краще проникають через гематоенцефалічний бар'єр;
- препаратів замісної терапії - не усувають причину фенілкетонурії, але підтримують нормальне функціонування організму (Леводопа спільно з Карбідофой, 5-оксітріптофан, 5-формілтетрагідрофолат);
- гепатопротекторів - підтримують функціонування печінки;
- протисудомних засобів;
- введення гена фенілаланінгідроксилази в печінку - експериментальний метод.
Особливості харчування новонароджених і дієтотерапія
На першому році життя дитини з ФКУ допустимо годування грудним молоком, але його кількість повинна бути обмежена. До 6 місяців допустимим рівнем споживання фенілаланіну є 60-90 мг на 1 кг ваги малюка (в 100 г молока міститься 5, 6 мг фенілаланіну). Починаючи з 3 місяців, раціон дитини слід поетапно розширювати, вводячи в нього фруктові соки і пюре.
Дітям з 6 місяців дозволено введення в раціон овочевих пюре, каш (з саго), безбілкових киселів. Після 7 місяців можна давати малюкові низькобілковий макаронні вироби, з 8 місяців - хліб, який не містить білка. Вік, до якого слід обмежувати надходження білка в організм хворої дитини, не встановлено. Лікарі до теперішнього часу дискутують з питання доцільності довічної дієтотерапії, але сходяться на думці, що мінімум до 18 років необхідно дотримуватися дієтичного харчування.
Фенілкетонурія, діагностована у жінки, не є приводом відмовитися від народження дитини. Майбутнім мамам з ФКУ для попередження пошкодження плоду під час вагітності та профілактики можливих ускладнень необхідно до планованого зачаття і під час виношування дитини дотримуватися дієти з обмеженням фенілаланіну (його рівень в крові повинен бути до 242 мкмоль / л).
Безлактозні суміші для малюків
Дієта при фенілкетонурії базується на суттєвому зменшенні дози натурального білка в добовому раціоні, але організм новонародженої дитини не може розвиватися нормально при відсутності необхідних мікроелементів. Для заповнення потреби малюка в білку застосовуються безлактозние амінокислотні суміші, якими, відповідно до російського законодавства, хворі повинні забезпечуватися безкоштовно.
Толерантність немовляти до фенілаланіну протягом першого року життя стрімко змінюється, тому необхідно контролювати його концентрацію в крові дитини і вносити коригування в дієту. Суміші розраховані на певні вікові групи:
- малюкам до року призначаються Афенілак 15, Аналог-СП, PKU-1, PKU-mix, PKU Anamix;
- дітям, старше 1 року, призначають збагачені вітамінами і мінералами суміші з підвищеним вмістом білка - PKU Prima, P-AM Universal, ФКУ-1, ФКУ-2, ХР Максамейд, ХР Максамум.

Дієтичні продукти для поповнення запасів білка
Одним з основних компонентів харчової дієти при фенілкетонурії є малобелковие продукти на основі крохмалю. Ці добавки містять гідролізат казеїну, триптофан, тирозин, метіонін, азот і забезпечують добову потребу організму дитини в білку, який необхідний для нормального розвитку і зростання. Спеціалізованими продуктами, заповнюють брак необхідних мінералів і амінокислот при їх нестачі в раціоні харчування, є:
- берлофен;
- Ціморган;
- Мінафен;
- Апонте.
Дієта для дітей дошкільного віку та школярів
У міру адаптації організму до фенілаланіну дітям з віку 5 років можна поступово зменшувати обмеження в харчуванні. Розширення раціону відбувається шляхом введення круп, молочних продуктів, м'ясних виробів. Школярі старших класів мають уже високу толерантність до фенілаланіну, тому в цьому віці можна продовжити розширення дієти, при цьому необхідно відслідковувати реакцію на всі зміни в харчуванні. Для контролю стану дитини застосовуються такі способи:
- оцінка неврологічних показників, психологічного стану;
- контроль показників електроенцефалограми;
- визначення рівня фенілаланіну.
Групи продуктів при ФКУ
У раціон харчування пацієнтів з ФКУ поряд з малобелковой крохмалистими продуктами і лікувальними сумішами входять і продукти натурального походження. При складанні меню слід чітко розраховувати кількість споживаного білка і не перевищувати рекомендовану лікарем дозування. Для виключення токсичного впливу на організм розроблені 3 списку продуктів, які містять заборонені (червоний), нерекомендовані (помаранчевий) і дозволені (зелений) позиції.
червоний список
Фенілкетонурія розвивається на тлі відсутності ферменту, що перетворює фенілаланін в тирозин, тому високий вміст білка є приводом для віднесення продуктів в заборонений (червоний) список. Позиції з цього переліку слід повністю виключити раціону харчування хворого ФКУ:
- м'ясо;
- внутрішні органи тварин, субпродукти;
- ковбаси, сосиски;
- морепродукти (в т.ч. риба);
- яйця всіх птахів;
- кисломолочні продукти;
- горіхи;
- плоди бобових і зернових культур;
- соєві продукти;
- желатінсодержащіе страви;
- кондитерські вироби;
- аспартам.
помаранчевий список
Продукти, які повинні дозовано надходити в організм дитини з діагнозом ФКУ, включені в помаранчевий список. Включення в раціон харчування позицій з цього переліку допустимо, але в строго обмеженій кількості. Ці продукти хоч і містять небагато білка, але теж можу підвищити рівень фенілаланіну, тому їх вживання не рекомендовано:
- консервовані овочі;
- страви з картоплі і рису;
- капуста;
- молоко;
- щербет.
зелений список
Безбілкові продукти дозволені до вживання хворими з діагнозом фенілкетонурія без обмежень. Перед покупкою позицій з зеленого списку необхідно вивчити склад, зазначений на упаковці, і переконатися, що там не міститься барвник аспартам, що містить фенілаланін:
- фрукти;
- овочі (за винятком картоплі та капусти);
- ягоди;
- зелень;
- крохмалисті крупи (саго);
- мед, цукор, варення;
- борошняні вироби з кукурудзяного або рисового борошна;
- масла, жири (вершкове, рослинне, оливкова).
Як контролювати рівень фенілаланіну в крові
Фенілкетонурія є невиліковним захворюванням, яке можна перевести в фазу стагнації шляхом застосування дієтотерапії та лікувально-профілактичних заходів. При зміні умов життя, порушенні режиму харчування хвороба може знову загостритися, тому хворим потрібно довічне спостереження. Процес контролю полягає в періодичному визначенні рівня фенілаланіну в крові. Частота здачі аналізів залежить від віку пацієнта:
- до 3 місяців - скринінг крові треба робити щотижня до отримання стійких результатів;
- від 3 місяців до 1 року - 1-2 рази на місяць;
- від 1 до 3 років - 1 раз за 2 місяці;
- старше 3 років - щоквартально.

Кров для аналізів здається через 3-4 години після прийому їжі. Крім скринінгу розвиток ФКУ контролюється шляхом визначення нутрітівного статусу, фізичного, емоційного розвитку хворого, рівня інтелектуальних здібностей і розвиненості мови. За результатами спостережень може виникнути необхідність додаткової діагностики із залученням відповідних фахівців.