
Спектр генодерматозов широкий. Деякі з них, такі як псоріаз, можуть протікати відносно легко та піддаватися лікуванню місцевими засобами. Інші, такі як іхтіоз типу арлекіно, можуть призвести до смерті. Нижче наведено п'ять спадкових проблем зі шкірою, які охоплюють більшу частину епідермісу.
Хвороба Дар'ї-Уайта
Хвороба Дар'є-Уайта, відома клінічно як фолікулярний кератоз, була вперше виявлена наприкінці 19 століття дерматологами Фердинандом-Жаном Дар'є та Джеймсом Кларком Уайтом. Уайт визначив розлад як спадкове захворювання шкіри, коли мати та дочка прийшли до нього на лікування.
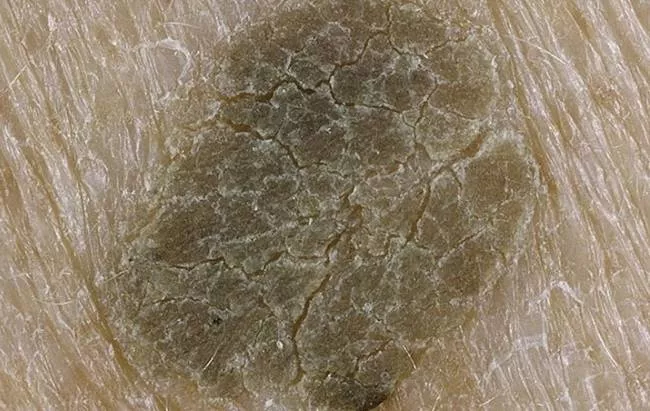
Хвороба Дар'є-Уайта характеризується аномальним затвердінням клітин шкіри на зовнішньому шарі шкіри процесом, званим зроговінням. Це той же процес, який перетворює здорові клітини шкіри на нігті, але у людей, які страждають на хворобу Дар'ї-Вайт, це відбувається в інших місцях шкіри. Дослідники виявили, що в клітинах шкіри пацієнтів відсутня сполука, яка зв'язує клітини разом (називається десмосоми), що може сприяти більш частому зроговенню.
Найпомітнішим симптомом є маленькі, жирні, жовто-коричневі, схожі на прищі папули, які масово утворюються в областях навколо сальних залоз.
Дослідники виявили зв'язок між мутаціями в гені ATP2A2 та хворобою Дар'є-Уайта, але також не виявили загального патерну мутацій, що визначає це захворювання. Навіть у людей з однаковими мутаціями ATP2A2 можуть виявлятися різні симптоми.
Бульозний епідермоліз
Бульозний епідермоліз (EB) - це виснажливе спадкове захворювання шкіри, яке призводить до утворення пухирів при незначних змінах тиску або температури. Зазвичай у людей виникає реакція на утворення пухирів, коли зовнішній та внутрішній шари шкіри відокремлюються. Вільна область заповнюється рідиною, яка служить подушкою, поки пошкоджена шкіра під нею гоїться.
Пацієнти з бульозним епідермолізом мають надзвичайно підвищену реакцію на появу пухирів. Проста ходьба, повзання, перебування на руках та/або навіть незначні зміни кімнатної температури можуть призвести до утворення хворобливих пухирів по всій шкірі. Частота утворення пухирів збільшує ймовірність зараження пацієнта інфекцією, що призводить до подальших ризиків для здоров'я, таких як ампутація.
Принаймні 10 різних генів були з'єднані в комбінації, щоб викликати EB. У більшості випадків захворювання передається у спадок від батьків, хоча воно також рідко може бути результатом випадкової мутації.
Найчастіше мутація в генах, відповідальних за експресію білків кератину, які забезпечують структуру та міцність зв'язку між шарами шкіри, успадковується від батьків.
Пластинчастий іхтіоз
Спадкове захворювання, пластинчастий іхтіоз, отримало другу половину своєї назви від латинського слова, що позначає рибу, їхтис. Цей термін підходить, оскільки у пацієнтів з пластинчастим іхтіозом в результаті їх захворювання по всьому тілу утворюються товсті лусочки.

У здорових людей старі клітини захищають молоді клітини перед випаданням та заміною. Під роговим шаром (зовнішній шар) клітини шкіри, відомі як кератиноцити, діляться утворюючи нові здорові клітини. У міру старіння та відмирання кератиноцити тверднуть і мігрують до рогового шару, утворюючи захисний бар'єр. Зрештою, вони випадають, оскільки їх замінюють знову затверділі клітини.
При пластинчастому іхтіозі клітини шкіри пацієнта розвиваються нормально, але в міру того, як вони тверднуть і мігрують у роговий шар, вони не відокремлюються, що запобігає їх випаданню. Зрештою, омертвілі клітини шкіри накопичуються і утворюють тверді лускаті пластинки, які покривають тіло і характеризують пластинчастий іхтіоз.
Захворювання зустрічається рідко. Як правило, у пацієнтів симптоми розвиваються в ранньому дитинстві або можуть народитися з лусочками. Симптоми можуть зникати протягом більшої частини дорослого життя і знову з'являтися пізніше в житті.
Кожна порфірія
Шкірні порфірії насправді складають шість різних типів спадкового захворювання порфірії. У кожній з них пацієнти не можуть виробляти ферменти, які створюють гем, компонент червоних кров'яних тілець, що доставляє кисень.
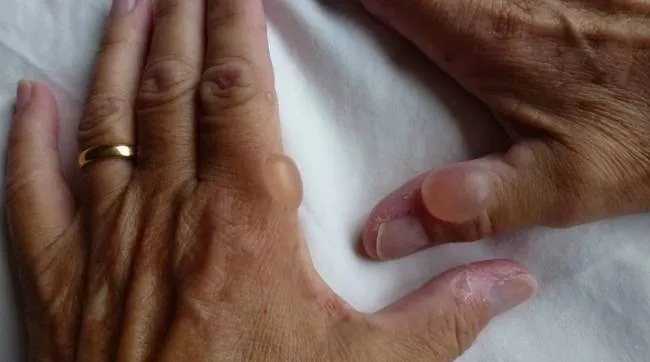
Гем складається з хімічних речовин, званих порфіринами, і вони можуть накопичуватися, якщо не перетворюються на гем. Нестача гему та накопичення порфіринів призводять до симптомів порфірії. Шкірні порфірії вражають шкіру.
У випадках шкірної порфірії шкіра пацієнта надзвичайно світлочутлива. Після дуже короткого перебування на сонці на шкірі з'являється почервоніння, хворобливе роздратування та пухирі. Шкіра також може набрякати під впливом сонячного світла та аномально темніти. У результаті пацієнтам настійно рекомендується уникати впливу сонячного світла.
Діти успадковують це захворювання від одного або кількох своїх батьків. Існує вісім різних ферментів, які перетворюють порфірини на гем, і спадкова мутація в будь-якому з генів, що експресують ці білки, може призвести до порфірії.
Кератодермія Маледа
Що стосується генодерматозів, хвороба де Маледа - одне із рідкісних типів. Це спадкове захворювання шкіри зустрічається в основному у людей середземноморського походження і було названо на честь острова Маледа, розташованого неподалік Хорватії, де були задокументовані перші випадки.
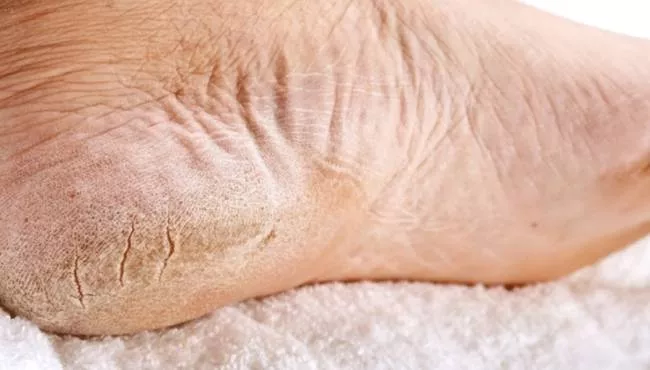
Захворювання є різновидом кератозу долонних і підошовних кісток, більш поширеного типу шкірного захворювання, що характеризується потовщенням шкіри на долонях рук і підошвах ніг. Це збільшення розмірів є результатом збільшення розмірів клітин шкіри. Це, у свою чергу, призводить до збільшення долонь і підошв, а також надає їм жовтуватого відтінку.
Оскільки хвороба де Маледа є рідкісним генодерматозом, не дивно, що це також аутосомно-рецесивне захворювання. Це означає, що необхідно внести дві копії гена, що мутував, по одній від кожного батька.
Ген SLURP1, який відповідає за кодування білків, що утворюють зв'язки між клітинами, був ідентифікований як винуватець хвороби де Маледа.
Увага! Інформація, подана у статті, має ознайомлювальний характер. Матеріали статті не закликають до самостійного лікування. Тільки кваліфікований лікар може поставити діагноз і дати рекомендації щодо лікування, виходячи з індивідуальних особливостей конкретного пацієнта.